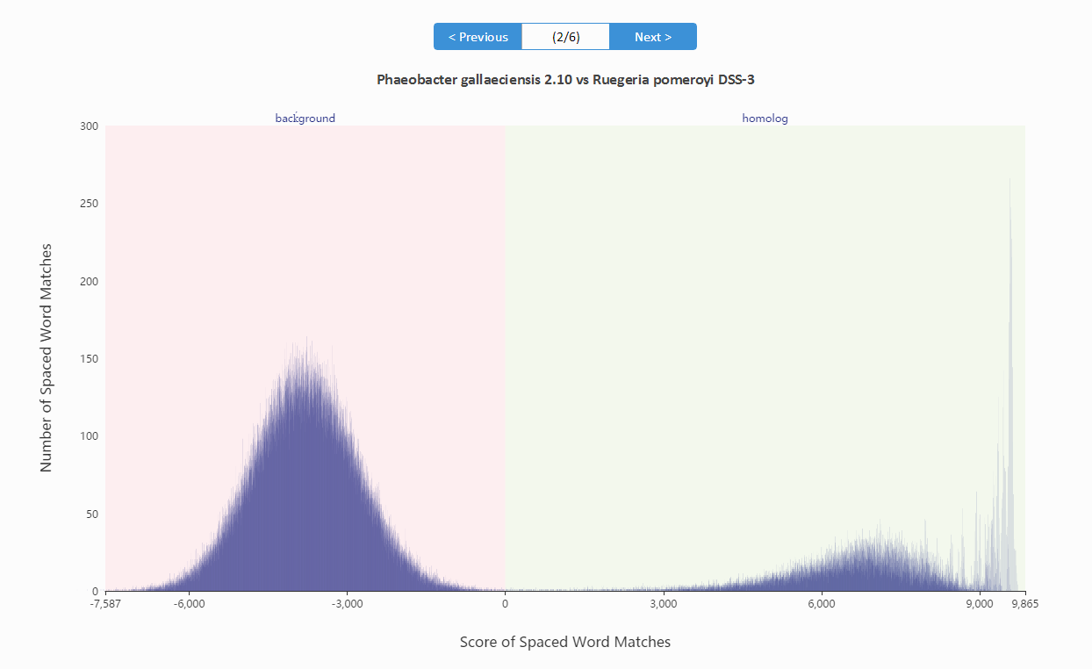
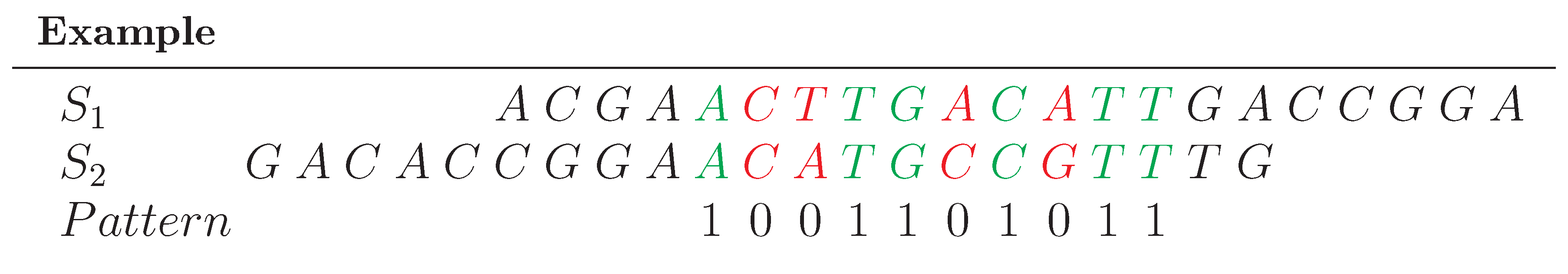For each pair of input sequences, the scores of the spaced-word matches can be visualized using a so-called 'spaced-word histogram'. Here, the number of spaced-word matches is plotted against the scores, i.e. for each possible score value the number of spaced-word matches with this score is plotted.
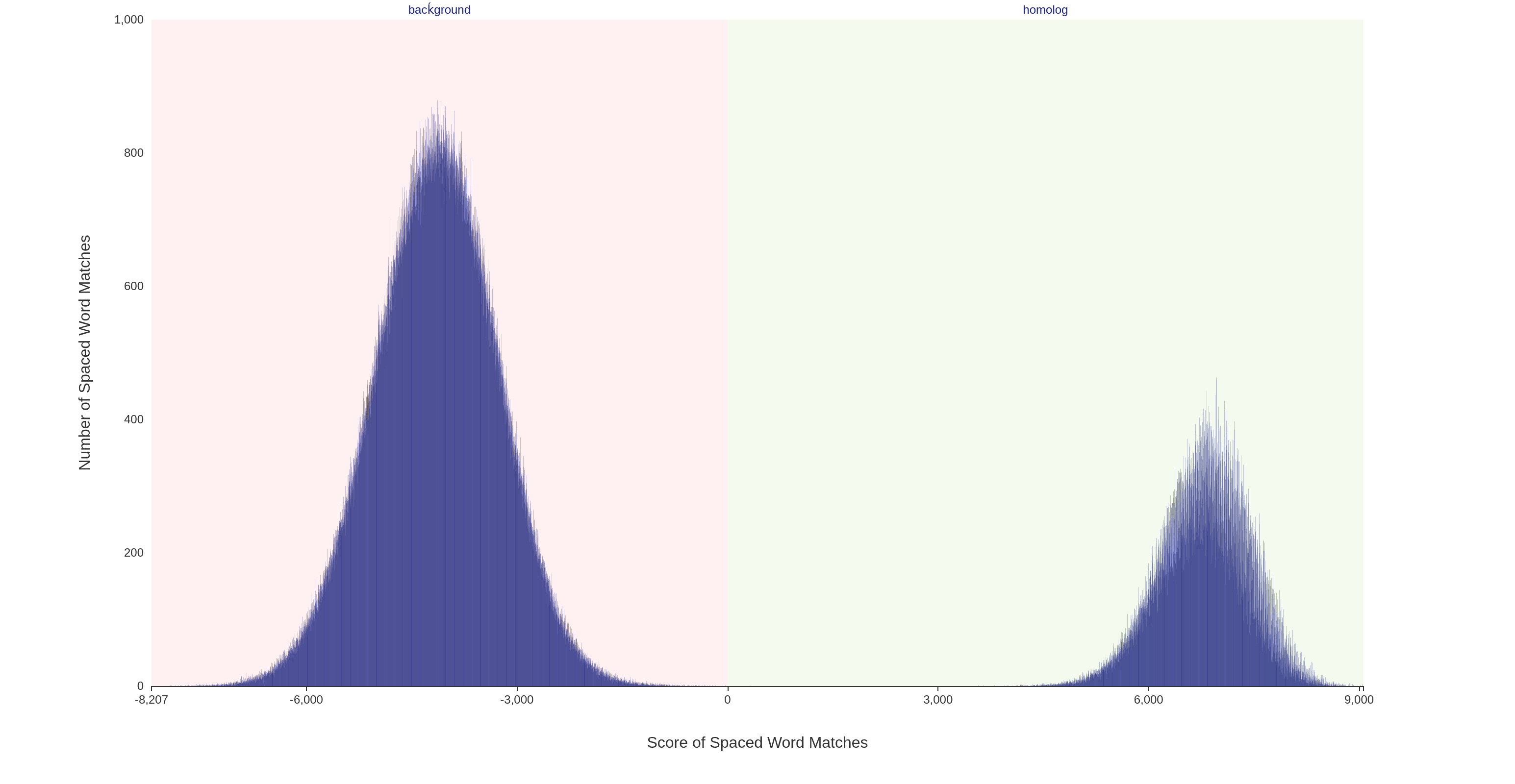
For a pair of i.i.d. random sequences, two peaks are visible in the spaced-word histograms which are both approximately binomially distributed (see above). The peak on the left-hand side (negative score values) essentially consists of random background hits, while the peak on the right-hand side (positive values) consists of spaced-word matches representing true homologies. Please note that for real data, the homologous peak looks more complex because the varying degrees of sequence similarity in different parts of the sequences, see below (Octadecabacter arcticus 238 vs Octadecabacter antarticus 307).
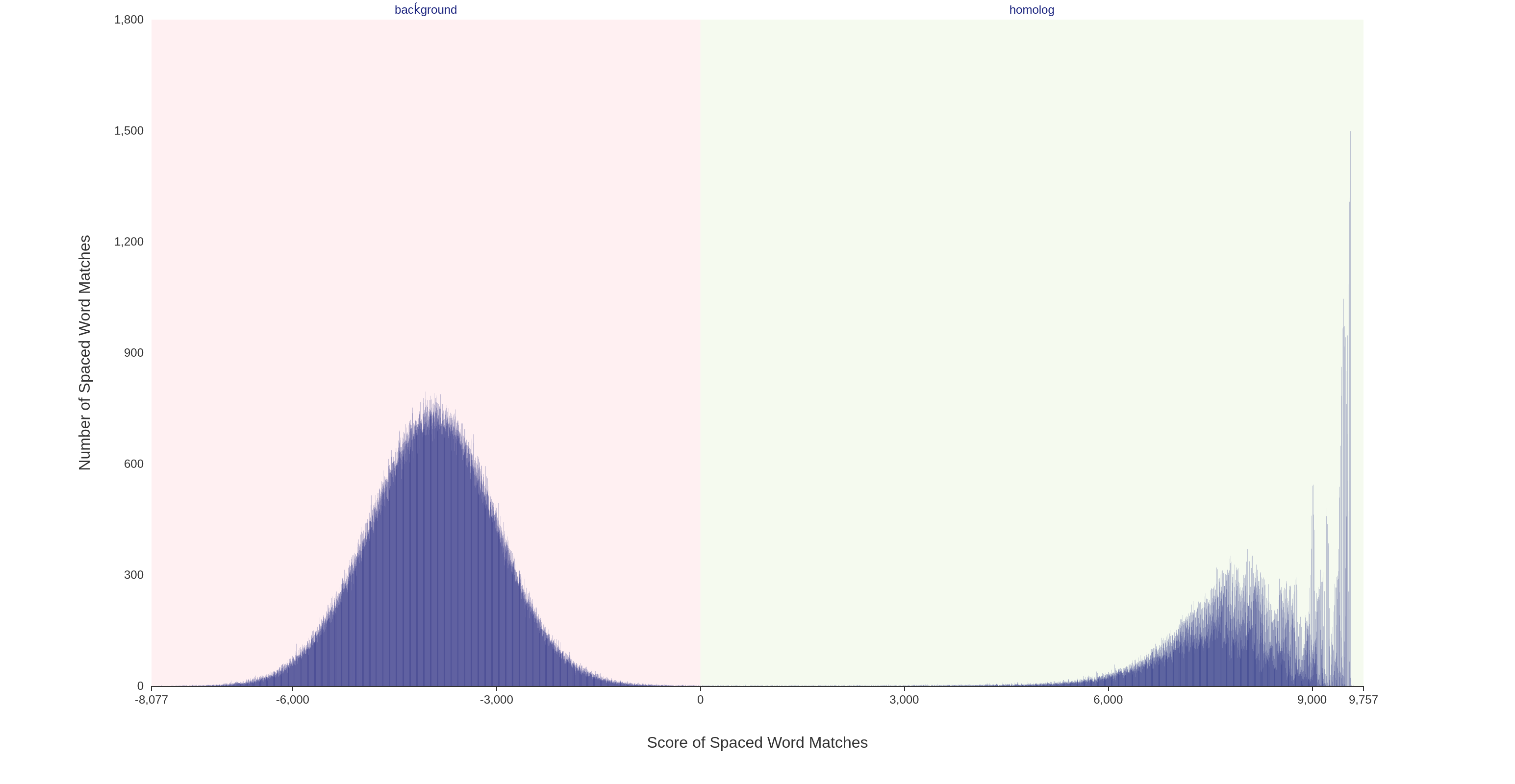
Spaced-word histograms can be visualized by our web server and the user can change the threshold for each pairwise sequence comparison individually, to distinguish true homologies from noise. Our command line tool, which can be downloaded in the download section, allows the user as well to change the threshold, but the difference compared to our web server is that only a global threshold can be set for all sequence pairs.
FSWM also removes ambigous spaced-word matches, see our paper for more details.